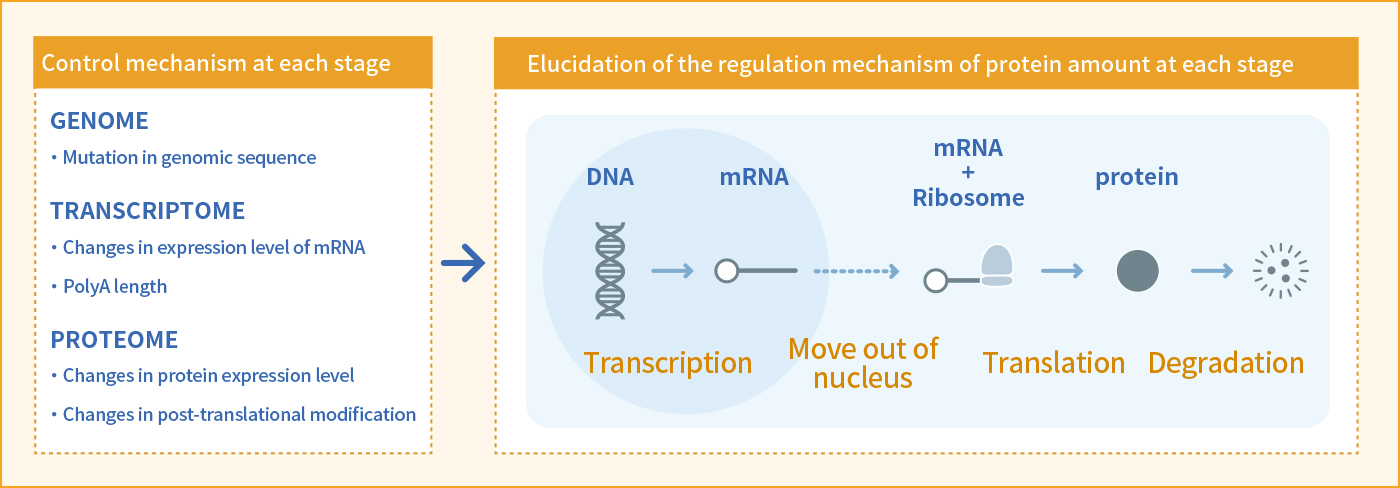
Cellular control mechanism by proteins
Central dogma proposed in 1958 by Dr. Crick, that gene expression is performed from transcribed mRNA (from DNA) to translated protein. This is still a basic principle of molecular biology.
However, the whole picture of what kind of control mechanism exists at each stage has not yet been elucidated. We analyze various large-scale data, identify genes whose amount of protein is specifically changing according to the cell types. We’d like to elucidate the control mechanism of amount of protein. If the regulatory mechanism of the protein that determines the identity of each cell is clarified, artificial control of cell fate could be possible. In addition, this research is important for a wide range of biology related fields such as pharmaceuticals and medical applications because we could uncover important proteins to target each cell types and to increase reprogramming efficiency.
However, the whole picture of what kind of control mechanism exists at each stage has not yet been elucidated. We analyze various large-scale data, identify genes whose amount of protein is specifically changing according to the cell types. We’d like to elucidate the control mechanism of amount of protein. If the regulatory mechanism of the protein that determines the identity of each cell is clarified, artificial control of cell fate could be possible. In addition, this research is important for a wide range of biology related fields such as pharmaceuticals and medical applications because we could uncover important proteins to target each cell types and to increase reprogramming efficiency.
The role of various post-translational modifications in stem cells
“Proteome analysis (proteomics)” is to study the complete set of proteins expressed in a cell or organism. In proteomics, a method using "mass spectrometer" connected to "high performance liquid chromatography" is broadly used. After fragmenting proteins into peptides, these peptides are separated by columns, and separated peptides are ionized and sequentially subjected to a mass spectrometer to identify and quantify the proteins.
Posttranslational modifications such as phosphorylation, methylation, ubiquitination, etc. play an important role not only in stem cells but also in various cell types. However, the amount of these post-translationally modified proteins were quite small and it is difficult to measure without enrichment. We will set up an analysis system that identify various kinds of modified proteins. Furthermore, we will uncover the effects of these post-translational modifications on stem cells and various differentiated cells.
Posttranslational modifications such as phosphorylation, methylation, ubiquitination, etc. play an important role not only in stem cells but also in various cell types. However, the amount of these post-translationally modified proteins were quite small and it is difficult to measure without enrichment. We will set up an analysis system that identify various kinds of modified proteins. Furthermore, we will uncover the effects of these post-translational modifications on stem cells and various differentiated cells.
Development of analytical and bioinformatics technologies
Along with the progress of mass spectrometers, efficiency of protein identification has drastically improved. However, identification of genes such as c-MYC and KLF4 is still difficult, which have important function in pluripotent stem cells but have low expression levels. Also, there are various methods to quantify proteins, but it is necessary to increase the quantification accuracy.
We will improve the analytical methods to increase the sensitivity to identify proteins with lower concentrations. We will also develop bioinformatics methods to improve quantitative accuracy, peak picking and precursor ion selections.
We will improve the analytical methods to increase the sensitivity to identify proteins with lower concentrations. We will also develop bioinformatics methods to improve quantitative accuracy, peak picking and precursor ion selections.