1. RNA二次構造予測のプログラム
ViennaRNAというUniversity of ViennaのLorenz先生、Hofacker先生が作られたプログラムです。結構アップデートをしてくれています。とてもありがたい。実行ファイルもダウンロードできるのですが、pythonとperl用の関数として使えるように設計してくれているものもあります。今回は、これまで使っていたperlではなく、pythonをそろそろ使えるようになろうと思って、anacondaで登録されているviennarnaパッケージをインストールしようと思いました。 しかし、ここで早くも問題が発生。MacとLinux版しかない。こういう研究している方々はみんなMac使いな印象ですが、こういうところに壁が現れます。幸運にも、Windows10からはLinuxが仮想環境で簡単に搭載できるようになっているらしく、まずはWindows10にLinux環境を整備することにしました。2. Windows10環境にLinuxを整備する
2A. Windows10の設定変更(Windows Subsystem for Linux (WSL)の有効化)
まずはWSLをWindows10上で有効にしないといけません。以下のサイトがとても分かりやすかったです http://www.aise.ics.saitama-u.ac.jp/~gotoh/HowToEnableWSL.html。次に”ubuntu”というLinuxをWindows10にインストールします。Ubuntuのインストールに関して、このサイトが一番わかりやすかったです(”rootユーザ”までのところを着実にやる)https://qiita.com/rubytomato@github/items/fdfc0a76e848442f374e。2B. ubuntu上、Windows上のファイルにアクセスする。
ubuntu上からWindows側にはcd /mnt/c/でCドライブにアクセスできます。Windowsからubuntu上のファイルには、エクスプローラのネットワークから\\wsl$という名前でアクセスすることができます。\\wsl$\Ubuntu\home\user名に行き、user名のフォルダを右クリックしてpropertiesを選んでAttributeのチェックを外しておきます。
3. anacondaのインストール
以下のサイトからlinux版anacondaをインストールする https://www.anaconda.com/products/individual。ダウンロードしたファイルをubuntuのuser名の下に移動して(エクスプローラーからドラッグアンドドロップしました)、bash Anaconda3-2020.02-Linux-x86_64をubuntuのコンソールから実行する(ファイル名は適宜変える)。参考にしたウェブサイトはここでした(問題が生じなければ”Anacondaのパスを通そう!”までで大丈夫)https://www.sejuku.net/blog/85373。
次に、anacondaにviennarnaを入れる環境を作ります。新しい環境を作らずにrootにパッケージを入れてしまって、かなり苦労したので、大元の環境はいじらないというのが鉄則かと思います。conda create -n py37 python=3.7 anaconda と打って、python=3.7という環境を作ります。これは、viennarnaの要求するpythonのversionが2020年5月28日時点では3.7だったためです。環境の作り方は以下のサイトを参考にしましたhttps://qiita.com/ozaki_physics/items/985188feb92570e5b82d。 新しい環境が本当に作成されたかconda info -eで確認します。そして、仮想環境をactivateします。このコマンドを打ちます。source activate py37 そして、もう一度conda info -eで確認して*が移っているのを確認します。これで準備OK。
4. viennarna, biopythonをインストールする
まずviennarnaが入っているチャンネルを登録します。conda config --add channels bioconda ともう一つ、conda config --add channels conda-forgeを登録します。そして一応、conda search viennarnaでviennarnaがあることを確認してから、conda install -c bioconda viennarnaでインストールします。そして、後でファスタ形式のファイルを使うのでbiopythonもインストールします。conda install biopythonこれで使える状態になっているはず。コンピューター側のセットアップ完了。
5. 5’UTR, 3’UTR, CDS, cDNAの配列を取得する
ここから、ちょっと話は変わるので、1-4を先にやっても、5を先にやってもどちらでも良いです。さて、翻訳されてタンパク質になるmRNAはどのような配列を持っているでしょうか。mRNA全体は逆転写するとcDNAの配列に相当し、5’末端から、5’UTR + CDS(タンパク質に翻訳される配列) + 3’UTR、となります。このそれぞれの配列をヒトのタンパク質種類全部欲しい。ここでとても便利なWEBサイトがEMBLのBioMartです。ほんと最高のWEBサイトです。詳しい説明は遺伝研の井上先生のページがとても参考になりました(特に”スプライシングされていない遺伝子配列を得る”のところ)。とにかく、5’UTRなどをとってこれます。個人的に「最高かよ」と思ったのは、以下の画像のmRNAの配列の図が出ているところです。5’UTRとか3’UTRとか選択を変えると、図の赤枠も変わります。ヒトであれば、この通りの設定をして、左上の”Result”を押して、右上の”Go”を押せば、ダウンロードされます。それぞれのテキストファイル(5’UTR, 3’UTR, CDS, cDNA)をダウンロードします。この左のFilterで”With CCDS ID(s): Only”を選んでいるのは、これまた私の好きなCCDSデータベースというのがNCBIにありまして、遺伝子ごとのmRNAの配列とタンパクの配列の代表を載せているデータベースです(ちょっと説明の仕方は自信ないです)。例えばNANOG遺伝子のCCDSの登録はこのようになっています。エキソン毎のmRNA配列と、それに対応するタンパク質配列が色分けで示されているところが、かなり好きなポイントです(エキソン境界の配列を見たいときあるじゃないですか)。話はそれましたが、CCDS IDのみを選んでいるのは、mRNAの配列の登録数は非常に多いので(ヒト遺伝子だと10万種類以上)、タンパク質と紐づいているmRNAの配列のみ(2万種類くらい)を取得したかったからです。これで、計算に必要な材料は手に入りました。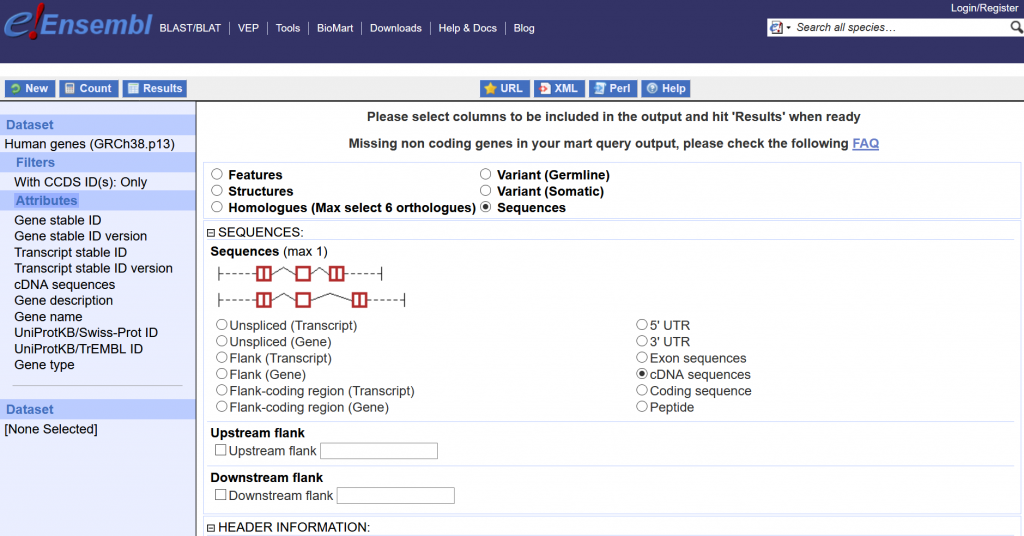
6. Pythonのプログラムを作って、RNA minimum free energyの計算結果を出力する
以下のスクリプトをRNAfold-calculate.pyというファイルに保存し(ファイル名は何でもいいけど)、ubuntuのホームディレクトリに置きます(場所も実行さえできればどこでもいいけど)。同じディレクトリに”Data”というフォルダを作り、その中に先ほどダウンロードした配列ファイルを保存します。そして、”Data”フォルダの中に”Result”というフォルダを用意してください。このスクリプトをpython RNAfold-calculate.pyで実行すると、結果ファイルは”Result”フォルダに出力されます。ただ、自分の愛用しているLet’s note (Core i7, 2.80 GHz, 16 GBメモリ)を使っても、ヒトの全遺伝子配列の計算結果が返ってくるのは2日後くらいかなと思います。Macを持っている場合はunix入ってるし、viennarnaもすんなり入るのでそんなに問題はないかと。
#############################################################
## to calculate the RNA minimum free energy using viennarna
import RNA
import re
import sys
from glob import glob
from Bio import SeqIO
from Bio.Alphabet import IUPAC
from Bio.Seq import Seq
files = glob("./Data/*")
for fname in files:
print(fname)
## output
output = fname
output = output.replace('.txt', '') + "-output.txt"
output = output.replace('./Data/', '')
output = './Data/Result/' + output
print(output)
with open(str(output), mode='w') as f:
print('ID', '\t', 'Minimum free energy', file=f)
# The RNA sequence from file
fasta_in = str(fname)
for record in SeqIO.parse(fasta_in, 'fasta'):
id_part = record.id
desc_part = record.description
seq = record.seq
dnaseq = Seq(str(seq), IUPAC.ambiguous_dna) ## array like
rnaseq = dnaseq.transcribe()
#print('id:', id_part)
#print('desc:', desc_part)
#print('seq:', seq)
(ss, mfe) = RNA.fold(str(rnaseq))
print(id_part,'\t',mfe, file=f)
print('mfe:', mfe)
f.close
#############################################################
この内容は実習とかインターンシップの課題とかで使えそうだな。